近日,材料学院马东伟副教授与电子科技大学孙旭平教授、四川师范大学李权教授等合作,通过先进的实验合成、表征和自旋极化第一性原理计算方法,报道了导电二维镁金属有机框架用于高效O2电还原生成H2O2的最新成果。迄今为止,过渡金属基材料是主要的2电子ORR催化剂,而作为生物系统重要组成部分的主族元素基催化剂在传统电催化过程中的催化性能鲜有报道。以包含主族元素Mg的Mg3(HITP)2为例,实验表征发现这类催化剂在−0.2 ~ 0.7 V范围内具有较高的H2O2选择性,达90%左右(图a-b)。相关成果以“Conductive Two-Dimensional Magnesium Metal−Organic Frameworks for High-Efficiency O2 Electroreduction to H2O2”发表在国际化学顶级期刊《ACS Catalysis》上。该杂志影响因子为13.084,JCR分区一区,论文链接:https://pubs.acs.org/doi/10.1021/acscatal.2c00819。
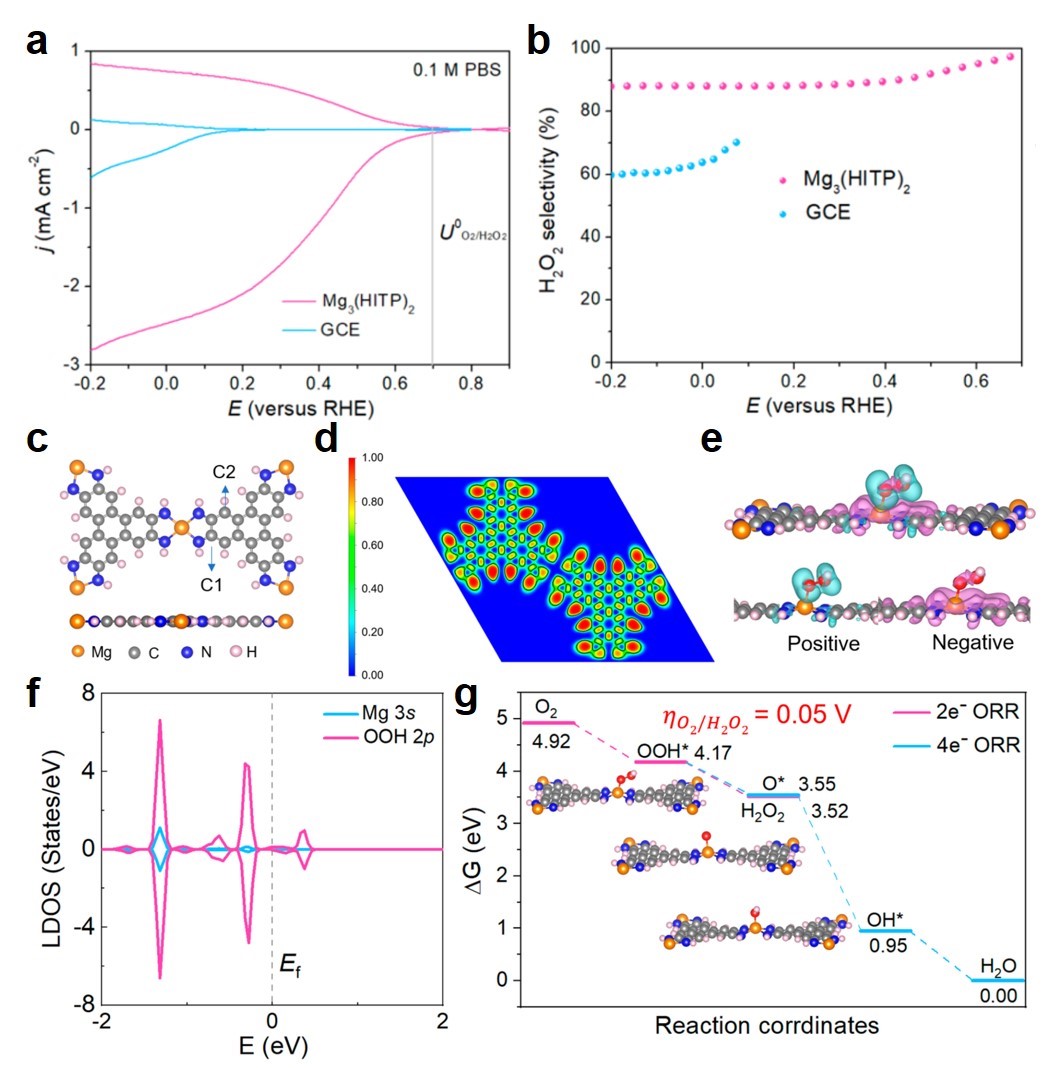
图 a,在0.1 MPBS中以1600 rpm记录Mg3(HITP)2和GCE的LSV曲线,以及分别在盘电极和环电极条件下测试的ORR和H2O2电流密度。b,通过a图计算得到的H2O2选择性。理论计算得到的c,Mg3(HITP)2原子结构;d,电子局域函数;e,电子差分密度;f,电子态密度;g,2电子ORR和4电子ORR反应自由能。
理论计算结果显示Mg处的电子局域几乎为零(图d),其与紧邻的N原子之间形成离子键,说明Mg以离子形式存在的方式作为活性中心,该结果与实验现象相吻合。Bader电荷分析和电子差分密度(图e)显示Mg的所有价电子都转移到周围原子上,说明Mg2+阳离子将非常有益于2电子ORR反应过程中关键中间体OOH*的吸附。计算的OOH*在Mg原子上的结合自由能是4.17 eV,其不仅比C1和C2位的结合更强,而且非常接近报道中具有最佳反应活性时的4.22 eV。电子态密度显示Mg与中间体OOH*以离子键相互作用(图f)。这些微观层次的理论计算结果都预示着Mg3(HITP)2的活性中心Mg2+对2电子ORR具有潜在的高活性。在通过详细计算2电子ORR反应过程生成H2O2的自由能后,发现该反应在Mg2+位的过电势非常低,仅为0.05 eV(图g)。尽管在Mg2+活性中心也可能发生4电子ORR反应生成H2O,但是理论计算结果表明Mg2+离子对O*的低亲和力使得2电子ORR比4电子ORR占据绝对的反应优势,即在实验中观察到的Mg2+离子基催化剂在−0.2 ~ 0.7 V范围内具有90%的高H2O2选择性。
该工作不仅为人们提供了一种可在环境条件下有效地将O2转化为H2O2的催化剂,还成功地开发了以主族元素为活性点的高效电催化剂,拓宽了含主族元素材料的应用领域。
电子科技大学孙旭平教授、河南大学材料学院马东伟副教授、四川师范大学李权教授是论文的共同通讯作者。该研究工作得到国家自然科学基金和河南省科技创新人才项目的资助。





 首页
首页



